Thermodynamique classique
Description macroscopique en thermodynamique
- 1h40min
- 23 inscrits
- Moyen
- Version du 06/08/2024
En thermodynamique classique, on s'intéresse à des systèmes composés d'un très grand nombre de particules.
A l'échelle macroscopique, l'expérience montre qu'on peut mesurer des grandeurs physiques (température, volume, pression, quantité de matière) qui n'ont de sens qu'à cette échelle, et qui suffisent à décrire complètement l'évolution du système.
Ces grandeurs ne jouent aucun rôle en mécanique. Pourtant, leur évolution relative semble obéir à des lois, selon les matériaux considérés. Dans le cas des gaz, il apparait même qu'à faible pression, ces lois deviennent universelles et ne dépendent plus de la nature des gaz considérés.
Dans ce cours, on va introduire un cadre conceptuel très général (système, variable d'état, fonction d'état, équation d'état) permettant de décrire tout système thermodynamique. En prenant l'exemple des gaz aux faibles pressions, on exhibera, à partir des lois empiriques historiques sur leur comportement thermoélastique, la loi du gaz parfait.
Ce que vous allez apprendre
- Définir un système en thermodynamique
- Différencier les systèmes ouvert, fermé, isolé
- Définir un variable, une fonction et une équation d'état
- Distinguer une grandeur extensive et une grandeur intensive
- Enoncer les postulats de la thermodynamique classique
- Comprendre le comportement asymptotique du gaz parfait
- Définir et donner l'intérêt des coefficients thermoélastiques
- Connaître les coefficients thermoélastiques pour un gaz parfait
- Etablir les coefficients thermoélastiques à partir d'une équation d'état
- Démontrer l'équation d'état du gaz parfait à partir des résultats empiriques des expériences historiques
- Etablir une équation d'état à partir de coefficients thermoélastiques
Comme en mécanique, en thermodynamique, on doit toujours, préalablement à toute analyse, choisir un système.
Système, Univers, extérieur
En thermodynamique:
On appelle système un ensemble comprenant un très grand nombre de particules (atomes, molécules, etc.), situé à l'intérieur d'une surface. La partie de l'Univers qui n'est pas le système est appelé l'extérieur.
Exemples: Une tasse de café, une voiture, une plante, un corps humain, une étoile, etc.
Système ouvert vs fermé
On dit que le système est fermé s'il n'échange pas de matière avec son extérieur ; on le dit ouvert dans le cas contraire.
Dans la suite, on se placera uniquement dans le cas de systèmes fermés : leur quantité de matière $n$ sera donc toujours constante.
Système isolé
On dit qu'un système est isolé lorsqu'il n'échange ni matière, ni énergie avec l'extérieur. On le dit non isolé dans le cas contraire.
Ainsi, un système isolé est nécessairement fermé. Attention, la réciproque est fausse !
En vertu du principe d'équivalence de Joule, l'énergie se transfère sous deux formes: un transfert mécanique (appelé travail) ou un transfert thermique (appelé chaleur).
L'expérience montre qu'on ne peut mesurer certaines grandeurs macroscopiques qu'à l'équilibre thermodynamique.
L'équilibre thermodynamique présuppose d'attendre suffisamment longtemps pour avoir un équilibre thermique (égalisation des températures du système avec son extérieur) et mécanique (égalisation des pression du système et de son extérieur).
Variable d'état
On appelle variable d'état une grandeur physique macroscopique, définie uniquement à l'équilibre, décrivant l'état du système.
Exemples:
| Symbole | Grandeur | Unité | Définition |
|---|---|---|---|
| p | pression | Pa | collision des particules |
| V | volume | m3 | espace exploré par les particules |
| T | température | K | agitation thermique des particules |
| n | quantité de matière | mol | nombre de particules |
Fonction d'état
On appelle fonction d'état une grandeur physique macroscopique dépendant exclusivement des variables d'état du système.
On déduit immédiatement qu'une fonction d'état n'est définie qu'à l'équilibre thermodynamique.
Ainsi, $H(S,V,p)=U(S,V)+p V$ est une fonction d'état, car elle dépend de l'énergie interne $U$ qui est une fonction d'état, et de deux variables d'état ($p$, $V$). En revanche, $H^{*}=U+p_{ext} V$ n'est pas une fonction d'état, car $p_{ext}$ n'est pas une variable d"état.
D'un point de vue mathématique, une fonction d'état est une fonction de plusieurs variables.
Un système thermodynamique étant constitué d'un grand nombre de particules, on répartit les grandeurs physiques mesurables (variables ou fonctions d'état) du système en deux catégories.
Grandeur extensive
Une grandeur extensive est proportionnelle à la quantité de matière du système.
Ainsi, si un même système double de "taille", la grandeur extensive double en valeur : $$V(2 \times n)=2 \times V(n)$$
Exemples: Masse $m$, volume $V$, quantité de matière $n$, énergie $E$, etc.
Grandeur intensive
Une grandeur intensive est indépendante de la quantité de matière du système.
Exemples: Pression $p$, température $T$, etc.
On remarquera que :
- une constante physique est, par construction, intensive
- le quotient de deux grandeurs extensives est intensif
- le quotient de deux grandeurs intensives reste intensif
Le concept de grandeur extensive/intensive fournit un nouveau critère de vérification qu'une relation physique n'est pas "complètement fausse" (avec l'homogénéité des dimensions).
$\frac{p}{T}=\frac{R}{V}$ où $R$ est une constante (intensive) n'est pas "homogène".
$\frac{p}{T}=\frac{nR}{V}$ où $R$ est une constante (intensive) est "homogène".
En thermodynamique classique, on postule :
(i) L'état thermodynamique d'un système à l'équilibre peut être décrit par un nombre restreint de variables d'état.
(ii) Lors d'une évolution entre 2 états thermodynamiques, la variation d'une fonction d'état ne dépend pas du chemin suivi.
Remarques:
a) Alors qu'en mécanique, pour décrire un grand nombre de particules il faudrait, pour chacune, disposer de leurs coordonnées et de leurs vitesses, la thermodynamique classique postule qu'il lui suffit d'un nombre très restreint de variables pour décrire les phénomènes physiques !
b) Les variables et fonctions d'état peuvent ne pas être connues ou même définies, entre deux états d'équilibre. Par contre, elles sont définies à chaque fois que l'état d'équilibre est atteint.
c) Pour ce qui concerne ce cours, on ne recherchera pas à connaître les états intermédiaires d'un système. Il s'agit néanmoins d'un domaine de recherche important (la thermodynamique hors équilibre).
d) Dans la suite, puisque le système est supposé fermé ($n=\textrm{constante}$), on travaillera uniquement avec 3 variables d'état : $p$, $T$, et $V$.
e) D'un point de vue mathématique, une fonction d'état est une fonction de plusieurs variables \textit{différentiable}. Par exemple, $V(T,p)$, lorsque $T$ passe à $T+dT$ et $p$ à $p+dp$ varie de $dV$ : $$dV=\frac{\partial V}{\partial T}\bigg|_{p} dT+\frac{\partial V}{\partial p}\bigg|_{T} dp$$
Au XVIII et XIX siècles, les physiciens ont mené des expériences sur des systèmes fermés constitués de gaz, permettant de connaître la variation relative de deux variables d'état, la troisième étant fixée.
L'une des 3 variables d'état étant fixée, il est commode de représenter les états d'équilibre dans des diagrammes à deux dimensions, appelés diagrammes d'état. On ne fournit ici que 2 exemples de diagrammes d"état.
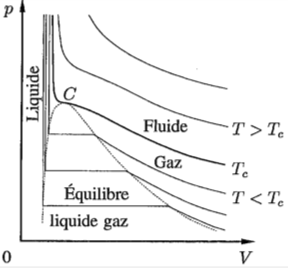
Dans le diagramme de Watt ($V$ en abscisses et $p$ en ordonnées) ci-dessus, ou diagramme $p-V$, on observe des courbes qui représentent des isothermes ($T$ fixée) d'Andrews.
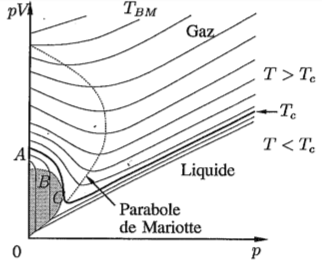
Dans le diagramme d'Amagat ($V$ en abscisses et $pV$ en ordonnées) ci-dessus, ou diagramme $pV-V$, on observe des courbes qui représentent des isothermes ($T$ fixée).
On peut distinguer plusieurs zones correspondant à des états physiques différents ; en particulier, les gaz possèdent un domaine d'existence vers les très faibles pressions.
Les diagrammes d'état précédents permettent de mettre en évidence qu'à faibles pressions ($p \longrightarrow 0$), pour les gaz, des comportements universels (qui ne dépendent pas de la nature du gaz considéré).
Par exemple, on peut rapprocher la loi de Boyle-Mariotte des diagrammes de Watt et d'Amagat.
Dans le diagramme $p-V$ (de Watt), lorsque $p$ devient proche de $0$, toutes les courbes, quelles que soient la température et la nature du gaz, tendent vers une hyperbole. Cela signifie que si $p \rightarrow 0$ alors $p \propto \frac{1}{V}$, la constante de proportionnalité dépendant de la température. Elle est même proportionnelle à la température.
Dans le diagramme $pV-V$ (d'Amagat), lorsque $p$ devient proche de $0$, toutes les courbes, quelles que soient la température et la nature du gaz, tendent vers une droite horizontale. Cela signifie que si $p \rightarrow 0$ alors $p V \approx \textrm{cste}$, la constante dépendant de la température. Elle est même proportionnelle à la température.
Ces constatations conduisent à la loi de Boyle-Mariotte:
On pourrait raisonner de façon similaire pour établir les lois de Charles et de Gay-Lussac, à partir d'autres diagrammes d'état.
Les coefficients thermoélastiques sont associés aux transformations particulières (pression, température ou volume constant) et expriment les variations relatives de l'une des variables d'état en fonction d'une deuxième variable d'état, la troisième étant fixée.
Du point de vue mathématique, il s'agit donc de quantités liées aux dérivées partielles des variables d'état ; physiquement, il s'agit précisément de "variations relatives entre deux variables d'état, toutes choses égales par ailleurs pour les autres variables d'état".
Coefficient de dilatation isobare
A pression constante, si la variation de la température de $T$ à $T+dT$ se traduit par une variation de volume de $V$ à $V+dV$, alors on définit le coefficient de dilatation isobare : $$\alpha=\frac{1}{V}\ \frac{\partial V}{\partial T}\bigg|_{p}$$
Physiquement, ce coefficient donne la variation relative du volume $\frac{\partial V}{V}$ lorsqu'on augmente la température $\partial T$, tout en maintenant la pression constante. Cette interprétation fournit une méthode expérimentale pour la mesurer ce coefficient pour n'importe quel système. Ce coefficient est donc lié à la pente en tout point $\frac{\partial V}{\partial T} \bigg|_{p}$ de la courbe $V(T)$ tracée dans le diagramme plan $(V,T)$, à $p$ fixée.
Ce coefficient est homogène à l'inverse d'une température et s'exprime en $\textrm{K}^{-1}$.
Pour les gaz à faibles pressions, on exploite la loi de Charles (1787), à $p=\textrm{cste}$ pour obtenir une relation différentielle.
\[ \begin{split} \frac{V_{1}}{T_{1}} = \frac{V_{2}}{T_{2}} &\Longleftrightarrow \frac{V}{T} = C(p) \implies V = C(p)T \implies \frac{\partial V}{\partial T}\bigg|_{p} = C(p) = \frac{V}{T} \\ \end{split} \]
D'où : \(\alpha = \frac{1}{V}\ \frac{\partial V}{\partial T}\bigg|_{p} = \frac{1}{T}\)
On divise par $V$ pour obtenir une variation relative et pour rendre intensive ce coefficient.
Coefficient de compression isotherme
A température constante, si la variation de la pression de $p$ à $p+dp$ se traduit par une variation de volume de $V$ à $V+dV$, alors on définit le coefficient de compression isotherme : $$\chi_{T}=-\frac{1}{V}\ \frac{\partial V}{\partial p}\bigg|_{T}$$
Physiquement, ce coefficient donne la variation relative du volume $\frac{\partial V}{V}$ lorsqu'on augmente la température $\partial T$ et qu'on maintient la pression constante. Cette interprétation fournit une méthode expérimentale pour mesurer ce coefficient pour n'importe quel système.
Ce coefficient est donc lié à la pente en tout point $\frac{\partial V}{\partial p} \bigg|_{T}$ de la courbe $V(p)$ tracée dans le diagramme plan $(V,p)$, à $T$ fixée.
Ce coefficient est homogène à l'inverse d'une pression et s'exprime en $\textrm{Pa}^{-1}$.
Pour les gaz à faibles pressions, on exploite la loi de Boyle-Mariotte (1662), à $T=\textrm{cste}$.
\[ \begin{split} p_{1} V_{1}=p_{2} V_{2} &\implies.p V= C(T) \implies V=\frac{C(T)}{p} \implies \frac{\partial V}{\partial p}\bigg|_{T}= -\frac{C(T)}{p^{2}} \\ \end{split} \]
D'où: $$\chi_{T}=-\frac{1}{V}\ \frac{\partial V}{\partial p}\bigg|_{T}=-\frac{1}{V} \bigg( -\frac{C(T)}{p^{2}} \bigg)=\frac{pV}{p^{2}V}=\frac{1}{p}$$
Coefficient de compression isochore
A volume constant, si la variation de la température de $T$ à $T+dT$ se traduit par une variation de pression de $p$ à $p+dp$, alors on définit le coefficient de compression isochore : $$\beta = \frac{1}{p}\ \frac{\partial p}{\partial T}\bigg|_{V}$$
Puisque $V(T,p)$ est une fonction d'état, la connaissance des coefficients thermoélastiques $\alpha$ et $\chi_{T}$ permet de remonter à l'expression analytique de la différentielle du volume $dV$.
Pour une fonction à une seule variable $V(n)$, on aurait : $$V(n+dn)-V(n)=dV=\frac{dV}{dn} dn=V'(n) dn$$
En transposant, pour une fonction de plusieurs variables $V(T,p)$: $$V(T+dT,p+dP)-V(T,p)= dV$$
avec: $ dV =\frac{\partial V}{\partial T}\bigg|_{p} dT+\frac{\partial V}{\partial p}\bigg|_{T} dp$
Donc:
\[ \begin{split} dV &= \alpha V dT - \chi_{T} V dp \\ &= V \left( \alpha dT - \chi_{T} dp \right) \\ \frac{dV}{V} &= \alpha dT - \chi_{T} dp \\ \end{split} \]
Les variables d'état d'un système sont \textit{a priori} indépendantes les unes des autres. Cependant, l'expérience permet, selon les matériaux considérés, de dégager certaines relations entre elles.
On appelle équation d'état d'un système thermodynamique une relation mathématique implicite impliquant exclusivement ses variables d'état : $$f(T,V,p)=0$$
Par construction, l'équation d'état n'est valide qu'à l'équilibre thermodynamique.
Sous certaines conditions, on peut formuler l'équation d'état sous forme explicite:
$f(T,V,p)=0$ \hspace{0.5cm} $\Longleftrightarrow$ \hspace{0.5cm} $V=g(T,p)$
On va s'intéresser à trois cas particuliers: le gaz parfait, les gaz réels "de Van der Waals" et les phases condensées (liquide, solide).
Puisque le comportement des gaz à faibles pression semble suivre des lois universelles qui ne dépendent pas de la nature du gaz considéré, on introduit le concept de gaz parfait.
Tous les gaz peuvent être modélisés par un gaz parfait pourvu que la pression soit suffisamment faible.
Les gaz vérifient alors l'équation d'état du gaz parfait: $$pV=nRT$$ également appelée couramment "loi du gaz parfait", due à Clapeyron.
Dans cette formule, $p$ est la pression (en Pa), $V$ est le volume en $\textrm{m}^{3}$, $T$ est la température (en K), $n$ est la quantité de matière (en mol). $R=8,314\ \textrm{J} \cdot \textrm{K} \cdot \textrm{mol}^{-1}$ est la constante du gaz parfait.
Cette équation d'état peut être démontrée et dérivée de plusieurs façons.
A partir de la différentielle totale de $V$:
\[ \begin{split} \frac{dV}{V} = \alpha \ dT-\chi_{T} \ dp &\Longleftrightarrow \frac{dV}{V} =\frac{1}{T} dT-\frac{1}{p} dp \\ &\Longleftrightarrow \frac{dV}{V} =\frac{dT}{T}-\frac{dp}{p} \\ &\Longleftrightarrow d \ln{V} +d \ln{p}= d \ln{T} \\ &\Longleftrightarrow d \ln{\big( pV \big)} =d \ln{T} \\ &\Longleftrightarrow \ln{\big( pV \big)} = \ln{T}+D(n)\\ &\Longleftrightarrow pV=C(n)T \\ \end{split} \]
A partir des coefficients thermoélastiques:
On intègre l'équation différentielle obtenue à partir de $\chi_{T}$:
\[ \begin{split} \chi_{T} = -\frac{1}{V} \frac{\partial V}{\partial p}\bigg)_{T} = \frac{1}{p} &\implies \frac{\partial V}{\partial p}\bigg)_{T}=-\frac{V}{p} \\ &\implies \frac{\partial V}{\partial p}\bigg)_{T}+\frac{V}{p}=0 \\ &\implies V(p,T)=D(T) e^{-\ln{p}} \\ &\implies V(p,T)=\frac{D(T)}{p} \\ \end{split} \]
La fonction $V(p,T)$ doit également vérifier l'équation différentielle obtenue à partir de $\alpha$. On injecte donc l'expression obtenue:
\[ \begin{split} \alpha = \frac{1}{V} \frac{\partial V}{\partial T}\bigg)_{p} = \frac{1}{T} &\implies \frac{\partial V}{\partial T}\bigg)_{p}=\frac{V}{T} \\ &\implies \frac{\partial }{\partial T}\bigg( \frac{D(T)}{p} \bigg)_{p}=\frac{V}{T} \\ &\implies D'(T)=\frac{pV}{T} \\ &\implies D'(T)-\frac{D(T)}{T} =0 \\ &\implies D(T)=C(n) e^{\ln{T}}=C(n) T \\ \end{split} \]
En combinant : $$V = \frac{C(n) T}{p}$$
Dans les deux cas, on parvient à : $pV=C(n) T$ ce qui confirme la loi d'Avogadro.
De plus, puisque $pV$ est extensif, $C(n)T$ doit l'être aussi. Puisque $T$ est intensif, $C(n)$ doit être extensif, c'est-à-dire $C$ est proportionnel à $n$. On décide de noter $C(n)=nR$, où $R$ est appelée constante du gaz parfait.
Finalement, on obtient bien la loi établie par Clapeyron :
$$pV = nRT$$
Lorsque la pression n'est plus proche de $0$, les gaz réels ne suivent plus l'équation d'état du gaz parfait.
Plusieurs équations d'état alternatives ont été proposées et sont encore affinées dans l'industrie aujourd'hui. On utilisera fréquemment l'équation d'état de Van der Waals:
$$\bigg( p+a\Big(\frac{n}{V}\Big)^{2} \bigg) \big(V-bn\big)=nRT$$
où le terme correctif $a \Big( \frac{n}{V} \Big)^{2}$ apparait comme une surpression et le terme correctif $bn$ a le rôle d'un volume inaccessible.
L'expérience montre que les phases condensées sont faiblement compressibles, c'est-à-dire :
$$\frac{\partial V}{\partial p}\bigg|_{T} \approx 0$$
par contre, ils peuvent être dilatables:
$$\frac{\partial V}{\partial T}\bigg|_{p} \neq 0$$
La surface en question peut être fermée (alors le nombre de particules est constant) ou ouverte (alors le nombre de particules peut varier).
On notera que les particules peuvent être sans masse (cas des photons) et que le système peut être dans tout état physique (solide, liquide, gaz, etc.), de sorte que la surface peut être rigide ou déformable.
Ainsi, la différence essentielle entre la description mécanique et la description thermodynamique résulte du nombre de particules prises en compte, de leurs interactions et, par conséquent, des grandeurs physiques permettant de décrire un système.
Par contre, un système fermé peut échanger de l'énergie (sous forme de chaleur ou de travail). Un système isolé est un système fermé qui, de plus, n'échange pas d'énergie. Il n'échange donc ni matière, ni énergie.
Par contre, pour chaque système thermodynamique étudié, il n'existe qu'un seul environnement. L'environnement est le complémentaire du système dans l'Univers. En conséquence, il existe une multitude d'environnement.
Une fonction d'état est une fonction des variables d'état. Par conséquence, elle n'est définie et mesurable qu'à l'équilibre du système.
Une équation d'état est une équation mathématique reliant plusieurs variables d'état entre elles. Elle n'est donc, elle aussi, valide qu'à l'équilibre.
Par contre, cette équivalence n'est pas toujours possible. Ainsi, on ne peut pas exprimer le volume $V$ d'un gaz de Van der Waals de façon analytique, en fonction de $p$, de $n$ et de $T$.
Le gaz parfait est un modèle qui représente bien n'importe quel gaz, pourvu que sa densité soit suffisamment faible (ou sa pression relativement faible ou son volume suffisamment grand). L'équation d'état est donc valide dans ces domaines extrêmes.
Le gaz de Van der Waals est un modèle qui représente bien n'importe quel gaz dans un domaine moins extrême de densité, pression ou volume. Il tient compte des interactions entre particules, au-delà des seules collisions.
Le gaz est représenté par les coefficients $a$ et $b$. De ce fait, l'équation de Van der Waals est moins "universelle" que celle du gaz parfait.
(i) L'état thermodynamique d'un système à l'équilibre peut être décrit par un nombre restreint de variables d'état.
(ii) Lors d'une évolution entre 2 états thermodynamiques d'équilibre, la variation d'une fonction d'état ne dépend pas du chemin suivi.
On peut voir le premier postulat comme la définition d'un état d'équilibre et le second postulat comme la définition d'une variable d'état. En d'autres termes, une variable d'état est une grandeur mesurable, macroscopique, définie à l'équilibre ... dont la variation ne dépend pas du chemin suivi. Si cette variation dépend du chemin suivi, alors il ne s'agit pas d'une variable d'état.
Au contraire, une grandeur intensive est indépendante de la quantité de matière du système.
On notera qu'une grandeur qui n'est pas intensive n'est pas nécessairement extensive, et réciproquement.
L'une des propriétés essentielles d'un coefficient thermoélastique est d'être intensif. De plus, on les définit positifs.
On le mesurerait donc en maintenant la pression constante, mais en faisant varier simultanément le volume et la température.
Pour obtenir son expression, on utilise les équations d'état :
(i) Pour le gaz parfait :
$$pV = nRT \implies p \frac{\partial V}{\partial T}\bigg|_{p} = nR \implies \alpha = \frac{1}{V} \frac{\partial V}{\partial T}\bigg|_{P} = \frac{nR}{pV} = \frac{1}{T}$$ (ii) Pour le gaz de Van der Waals :
$$\bigg( p+a\Big(\frac{n}{V}\Big)^{2} \bigg) \big(V-bn\big) =n RT \implies \bigg( p- 2 a \frac{n^{2}}{V^{3}} \frac{\partial V}{\partial T}\bigg|_{p} \bigg) \big(V-bn\big) + \bigg( p+a\Big(\frac{n}{V}\Big)^{2} \bigg) \frac{\partial V}{\partial T}\bigg|_{p} = nR$$ puis on isole $\frac{\partial V}{\partial T}\bigg|_{p}$ avant d'exprimer $\alpha$ : $$p \big(V-bn\big) - 2 a \frac{n^{2}}{V^{3}} \frac{\partial V}{\partial T}\bigg|_{p} \big(V-bn\big) + \bigg( p+a\Big(\frac{n}{V}\Big)^{2} \bigg) \frac{\partial V}{\partial T}\bigg|_{p} = nR$$ $$ \frac{\partial V}{\partial T}\bigg|_{p} = \frac{nR - p \big(V-bn\big) }{p + a\Big(\frac{n}{V}\Big)^{2} - 2 a \frac{n^{2}}{V^{3}} \big(V-bn\big) }$$ $$\alpha = \frac{1}{V} \frac{\partial V}{\partial T}\bigg|_{P} = \frac{1}{V} \frac{nR - p \big(V-bn\big) }{p + a\Big(\frac{n}{V}\Big)^{2} - 2 a \frac{n^{2}}{V^{3}} \big(V-bn\big) }$$ Dans les deux cas, on peut également calculer $\frac{\partial T}{\partial V}\bigg|_{p}$ puis inverser pour trouver $\alpha$.
On le mesurerait donc en maintenant le volume constant, mais en faisant varier simultanément la température et la pression.
Pour obtenir son expression, on utilise les équations d'état :
(i) Pour le gaz parfait :
$$pV = nRT \implies V \frac{\partial p}{\partial T}\bigg|_{V} = nR \implies \beta = \frac{1}{p} \frac{\partial p}{\partial T}\bigg|_{V} = \frac{nR}{pV} = \frac{1}{T}$$ (ii) Pour le gaz de Van der Waals :
$$\bigg( p+a\Big(\frac{n}{V}\Big)^{2} \bigg) \big(V-bn\big) =n RT \implies \frac{\partial p}{\partial T}\bigg|_{V} \big(V-bn\big) = nR \implies \beta = \frac{1}{p} \frac{\partial p}{\partial T}\bigg|_{V} = \frac{1}{p} \frac{nR}{V-bn}$$ On vérifie bien que ces formules coïncident lorsque $V \longrightarrow + \infty$
On le mesurerait donc en maintenant la température constante, mais en faisant varier simultanément le volume et la pression.
Pour obtenir son expression, on utilise les équations d'état :
(i) Pour le gaz parfait :
$$pV = nRT \implies V = \frac{nRT}{p} \implies \frac{\partial V}{\partial p}\bigg|_{T} = - \frac{nRT}{p^{2}} \implies \chi_{T} = - \frac{1}{V} \bigg( - \frac{nRT}{p^{2}} \bigg) = \frac{nRT}{Vp^{2}} = \frac{1}{p}$$ (ii) Pour le gaz de Van der Waals :
$$\bigg( p+a\Big(\frac{n}{V}\Big)^{2} \bigg) \big(V-bn\big) = n RT \implies p = \frac{nRT}{V-bn}-a \bigg( \frac{n}{V} \bigg)^{2} \implies \frac{\partial p}{\partial V}\bigg|_{T} = - \frac{nRT}{\big( V-bn \big)^{2}}+2a\frac{n^{2}}{V^{3}}$$ d'où: $$\chi_{T} = -\frac{1}{V} \frac{\partial V}{\partial p}\bigg|_{T} = -\frac{1}{V} \frac{1}{- \frac{nRT}{\big( V-bn \big)^{2}}+2a\frac{n^{2}}{V^{3}}}=\frac{1}{V}\frac{1}{\frac{nRT}{\big( V-bn \big)^{2}}-2a\frac{n^{2}}{V^{3}}} \underset{V \longrightarrow + \infty}{\longrightarrow} \frac{V}{nRT} = \frac{1}{p}$$